
The Future of Protein Modeling: AlphaFold and RoseTTAFold!
Explore the transformative impact of AlphaFold and RoseTTAFold on protein structure prediction. Discover their key features, methodologies, strengths, applications, and how their collaboration could shape the future of computational biology.
AI/FUTUREEDITOR/TOOLSA LEARNINGHEALTH/DISEASE
Sachin K Chaurasiya
1/23/20255 min read


Protein structure prediction is one of the most challenging and critical problems in biology. Understanding the three-dimensional (3D) structure of proteins is key to unlocking numerous advancements in medicine, drug discovery, and biotechnology. Two leading tools have emerged as game-changers in this field: AlphaFold by DeepMind and RoseTTAFold by the University of Washington's Baker Lab. Both systems have drastically improved the accuracy and efficiency of predicting protein structures, but they take different approaches and have unique strengths.
In this article, we explore the key features, methodologies, and impact of AlphaFold and RoseTTAFold, offering a comparative analysis to help you understand their significance and differences.
What is AlphaFold?
AlphaFold is an artificial intelligence (AI) system developed by DeepMind, a subsidiary of Alphabet Inc. It gained global attention after winning the Critical Assessment of Techniques for Protein Structure Prediction (CASP) competition in 2020. AlphaFold revolutionized protein structure prediction by solving structures with near-experimental accuracy.
Key Features
Deep Learning Approach: AlphaFold uses a deep learning model trained on vast amounts of protein sequence and structure data, including datasets from sources like the Protein Data Bank (PDB) and UniProt. These databases provide structural and functional information that helps AlphaFold learn the intricate relationships between amino acid sequences and their 3D conformations. It employs attention mechanisms and geometric learning to infer spatial relationships between amino acids.
End-to-End Prediction: AlphaFold’s model predicts the 3D structure of proteins directly from their amino acid sequences without relying heavily on templates or prior structural data.
High Accuracy: AlphaFold demonstrated accuracy comparable to experimental methods like X-ray crystallography or cryo-electron microscopy in many cases.
Open Access: In 2021, DeepMind released the AlphaFold Protein Structure Database, which now contains over 200 million protein structures, providing a transformative resource for researchers worldwide.
Integration with Tools: AlphaFold’s predictions are increasingly integrated into software pipelines for molecular dynamics simulations, aiding in the study of protein dynamics and interactions.
Strengths
Unprecedented Precision: It has set a new benchmark for accuracy in computational biology.
Broad Utility: AlphaFold’s predictions have been used in diverse fields, from understanding diseases to developing vaccines and enzymes for industrial applications. For example, it has been instrumental in studying the structure of SARS-CoV-2 proteins during the COVID-19 pandemic, enabling faster vaccine development. In biotechnology, AlphaFold has aided in designing enzymes for sustainable biofuel production and improved industrial processes, showcasing its transformative impact across disciplines.
Community Impact: Its open-access model democratizes protein structure data, empowering researchers globally.
Improving Biomedical Research: It aids in identifying genetic mutations’ impact on protein folding, enhancing the understanding of genetic diseases.
Challenges and Limitations
Resource-Intensive: Requires significant computational power and memory for large-scale predictions.
Complex Scenarios: Struggles with modeling protein-protein interactions or dynamics.
Static Predictions: Outputs are static structures, which may not capture dynamic conformational changes.


What is RoseTTAFold?
RoseTTAFold, developed by David Baker’s lab at the University of Washington, is another powerful AI-based protein structure prediction tool. It emerged shortly after AlphaFold and employs a different architectural approach inspired by natural language processing (NLP) models.
Key Features
Three-Track Network: RoseTTAFold integrates sequence, distance, and coordinate information through a novel three-track network architecture, akin to a three-lane highway where each lane (sequence, distance, and coordinate data) flows independently yet interacts dynamically at intersections to optimize the overall understanding of protein structures. allowing simultaneous learning of relationships across different dimensions.
Modular and Flexible: Unlike AlphaFold’s end-to-end design, RoseTTAFold’s modularity makes it more adaptable to specific use cases, such as modeling protein-protein interactions or designing novel proteins.
Computational Efficiency: RoseTTAFold is less computationally demanding than AlphaFold, requiring fewer GPUs and less memory, making it more accessible to researchers with limited computational resources. For example, while AlphaFold often needs high-end hardware like multiple GPUs for efficient processing, RoseTTAFold can run effectively on a single GPU with moderate specifications, significantly lowering the barrier to entry.
Open Source: The Baker Lab released RoseTTAFold as open-source software, encouraging customization and collaboration.
Improved Speed: RoseTTAFold’s algorithms enable faster predictions for certain tasks, such as multi-protein interactions.
Strengths
Versatility: Its modular design excels in scenarios requiring flexible applications, such as multi-protein complexes or de novo protein design.
Lower Resource Requirements: Its computational efficiency enables broader adoption across labs with limited infrastructure.
Innovative Architecture: The three-track network is a novel contribution to the field of protein modeling.
Protein Interaction Modeling: It is particularly strong in predicting and modeling interactions in protein complexes.
Challenges and Limitations
Lower Accuracy: Predictions may be less precise than AlphaFold for certain single-protein structures.
Data Dependence: Relies heavily on high-quality input data for optimal performance.
Limited Database: While open-source, it does not provide a comprehensive precomputed database like AlphaFold.
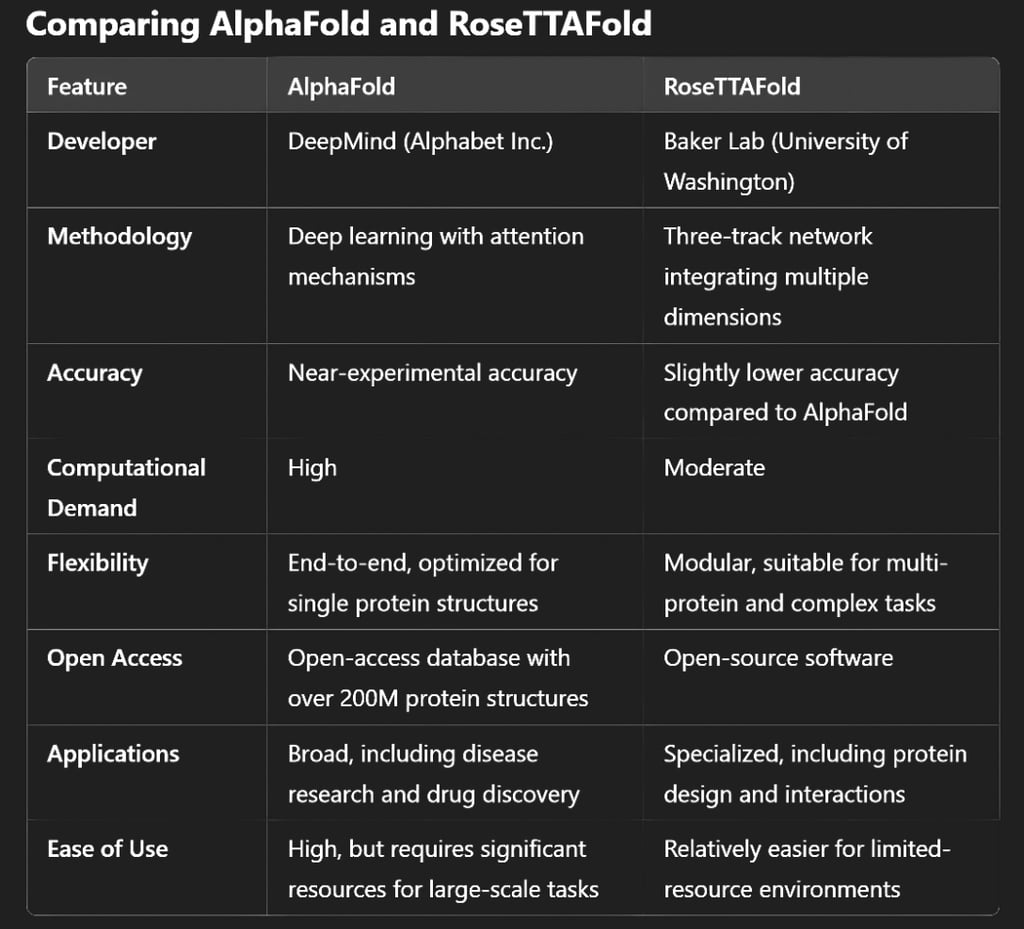
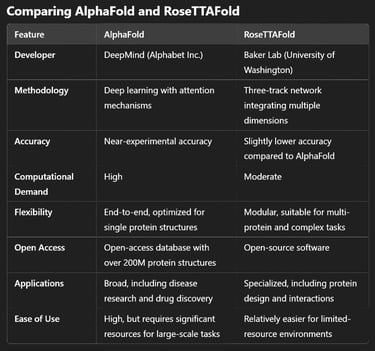
Applications and Use Cases
Drug Discovery
AlphaFold: Predicts drug targets with high accuracy, accelerating the development of therapies.
RoseTTAFold: Helps design novel therapeutic proteins, including antibodies and enzymes.
Fundamental Biology
AlphaFold: Enables researchers to study protein structures that were previously elusive.
RoseTTAFold: Facilitates understanding of protein interactions and novel complex formations.
Biotechnology
AlphaFold: Supports industrial enzyme design for biofuels, detergents, and more.
RoseTTAFold: Excels in designing synthetic proteins for specific functions.
Structural Genomics
AlphaFold: Its comprehensive database aids structural genomics projects by filling gaps in protein structure knowledge.
RoseTTAFold: Its adaptability helps researchers tackle unique challenges in genomic data analysis.
Future Prospects
The future of protein structure prediction will likely involve collaboration and integration between these tools. For instance, researchers could use AlphaFold’s high-precision predictions as a foundation and then apply RoseTTAFold’s modular architecture to explore protein interactions or multi-protein complexes. Hypothetically, hybrid pipelines could be developed where both tools are leveraged sequentially or in tandem to provide a more comprehensive analysis, addressing both static structures and dynamic interactions. Such synergy could revolutionize areas like drug discovery, synthetic biology, and systems biology by combining their strengths. Researchers may combine AlphaFold’s precision with RoseTTAFold’s flexibility to tackle even more complex biological problems. Additionally, advancements in computing power, algorithm optimization, and hybrid models will further enhance their capabilities.
Other areas of future exploration include:
Dynamic Modeling: Predicting protein dynamics and conformational changes in real time.
Integration with Experimental Methods: Using these tools to complement experimental techniques like cryo-EM and NMR.
Multi-Protein Complex Prediction: Developing hybrid approaches to improve the modeling of entire protein complexes.
Enhanced Accessibility: Efforts to make these tools more accessible to smaller labs and underfunded research institutions.
AlphaFold and RoseTTAFold represent monumental strides in computational biology. While AlphaFold excels in accuracy and single-protein prediction, RoseTTAFold shines in flexibility and efficiency. Together, they have redefined what is possible in protein structure prediction, driving progress in medicine, biotechnology, and fundamental research.
Choosing between the two depends on the specific requirements of a project. For high-precision single-protein modeling, AlphaFold is the ideal choice. For scenarios requiring flexibility, such as protein design or interaction studies, RoseTTAFold offers unique advantages. Ultimately, both tools are invaluable assets in the quest to understand and manipulate the molecular machinery of life.
Subscribe to our newsletter
All © Copyright reserved by Accessible-Learning
| Terms & Conditions
Knowledge is power. Learn with Us. 📚

